L’interessamento renale in corso di gammopatie monoclonali è molto variabile e può essere distinto in tre categorie:
- Danno renale in corso di mieloma multiplo. È la più frequente causa di danno renale in corso di gammopatia monoclonale. Nella nostra personale casistica, il danno renale acuto si osserva nel 20% dei pazienti affetti da mieloma multiplo, in linea con quanto descritto in letteratura (1). Nella maggioranza dei casi, si assiste alla deposizione di catene leggere nel tubulo distale con flogosi intra e peritubulare, che causano il quadro clinico istologico noto come “rene da mieloma” o “cast nephropathy”. Tuttavia, tutte le strutture renali possono essere coinvolte dal danno da proteine monoclonali, generando un ampio spettro di presentazioni clinico-patologiche.
- Danno renale in corso di malattia linfoproliferativa maligna (es. linfomi a cellule B). In rari casi, la gammopatia monoclonale è generata da un clone di B cellule producente, generalmente immunoglobuline di tipo IgM. Il quadro istologico, come nei pazienti affetti da mieloma multiplo, è proteiforme (2,3).
- Danno renale in corso di gammopatia monoclonale di significato non maligno (MGRS). È nota come gammopatia monoclonale di significato renale, ovvero una condizione in cui il clone plasmacellulare o di B cellule non presenta caratteristiche di malignità nonostante la presenza di un nesso patologico tra proteina monoclonale e danno tissutale (4) (tabella 1).
|
| MGUS | Smoldering Myeloma | Multiple Myeloma |
| M-Spike | <3 g/dl | ≥3 g/dl | ≥3 g/dl |
| Bone marrow PC% | <10% | ≥10% | ≥10% |
| Hypercalcemia | – | – | +/- |
| Renal impairment | – | – | +/- |
| Anemia | – | – | +/- |
| Osteolytic lesions | – | – | +/- |
Tabella 1
In tutte le categorie prima citate, l’eziopatogenesi è direttamente associata alla proteina monoclonale che per caratteristiche chimico-fisiche è causa di danno strutturale renale (vasi, glomeruli, tubuli, interstizio) (3). A fini diagnostici e prognostici, la biopsia renale ha importanza miliare, se praticabile in sicurezza (5).
Workup
- Kidney biopsy
- IF
- IHC
- Proteomics
- Find the source
- Bone marrow
- Lymphatic tissue
Infatti, solo essa può dimostrare l’interazione diretta tra il danno renale e la presenza della proteina monoclonale nei depositi tissutali (figura 1).
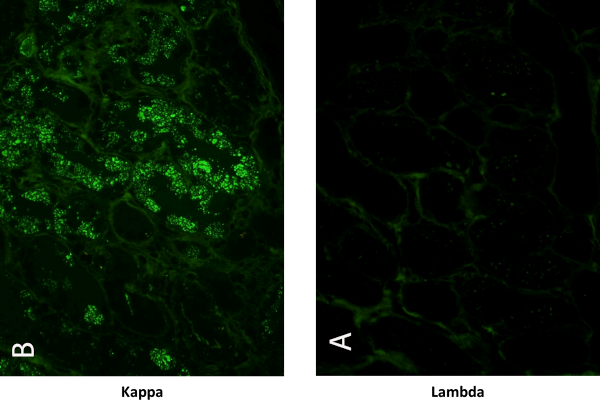
Figura 1
Accade inoltre che, seppur in presenza di depositi di proteine monoclonali, la sede e le caratteristiche fenotipiche del clone cellulare possono rimanere indeterminate (figura 2).
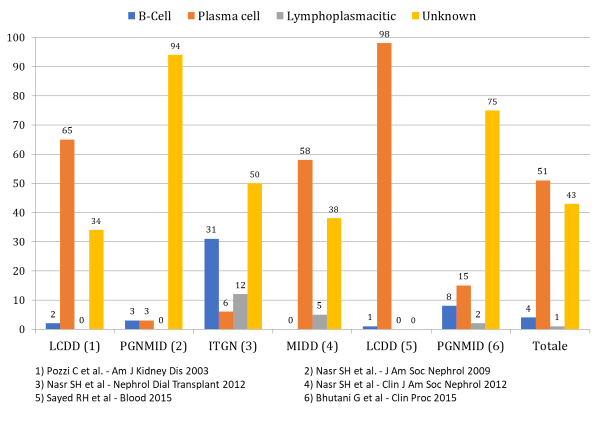
Figura 2
Quadri istologici più rilevanti:
- Cast nephropathy o “rene da mieloma”. È caratterizzata principalmente dalla deposizione di catene leggere monoclonali e proteina di Tamm-Horsfall nel tubulo distale, che insieme formano una matrice pro-infiammatoria intraluminare che è causa di danno tubulare acuto. L’impatto di questa condizione è prevalentemente renale.
- Amiloidosi primitiva (ad immunoglobuline, Ig). In generale, per amiloidosi si intende la deposizione tissutale di fibrille del diametro compreso tra 9 e 12 nm composte da monomeri di proteine la cui struttura terziaria è mutata. In corso di paraproteinemia, le fibrille di amiloide sono composte da monomeri di catene leggere (amiloidosi AL) o pesanti (AH) monoclonali. La sede di deposizione di fibrille di amiloide in sede renale (extra o intracellulare nelle cellule del tubulo prossimale, vascolare, interstiziale, glomerulare) caratterizza il quadro clinico: in particolare si può osservare sindrome di Fanconi se è presente coinvolgimento tubulare prossimale, insufficienza renale in assenza di proteinuria glomerulare se il coinvolgimento è interstiziale e/o vascolare, sindrome nefrosica se glomerulare, fino ad arrivare a quadri misti se la deposizione coinvolge più strutture renali. A livello sistemico, l’amiloidosi Ig causa interessamento multiorgano, in particolare cardiaco (frequente), gastroenterico, epatico, cutaneo e di altri tessuti.
- Malattia da Deposizione di Immunoglobuline Monoclonali (MIDD). È causata dalla deposizione di immunoglobuline monoclonali in sede prevalentemente glomerulare e del tubulo prossimale. In sede glomerulare è causa di una reazione mesangiale nodulare a depositi monotipici di catene leggere (light chain deposition disease, LCDD), pesanti (heavy chain deposition disease, HCDD), o entrambe (light and heavy chain deposition disease, LHCDD). In sede tubulare prossimale si osservano depositi monotipici a carico della membrana basale tubulare di tipo Randall.
- Glomerulonefrite proliferativa a depositi monoclonali di IgG (PGNMID). Questa rara entità è determinata dalla deposizione di IgG monoclonali (spesso kappa) in sede glomerulare, mimando una glomerulonefrite primitiva in assenza delle caratteristiche prima descritte nelle MIDD. I depositi sono esclusivamente glomerulari. Spesso il clone non è rilevabile in sede linfonodale o midollare, così come può non essere dimostrata una proteina monoclonale in sede urinaria o sierica.
- Alterazioni della via alterna del complemento. Una proteina monoclonale può agire come stabilizzatrice della C3 convertasi della via alterna o come inibitore di un fattore della regolazione della via alterna (es. inibitore del fattore H) (figura 3).
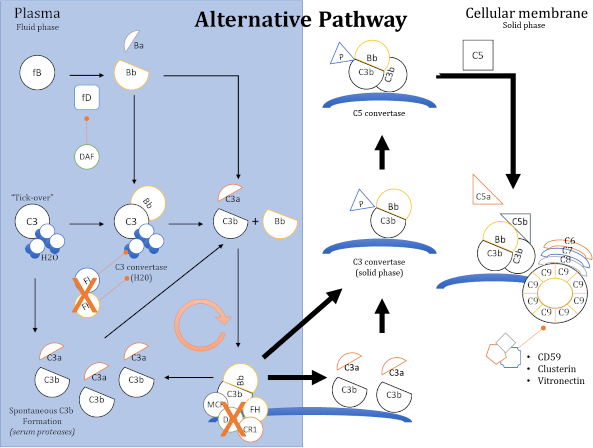
Figura 3
In relazione alla porzione di via alterna alterata (fase fluida o fase solida) potremmo osservare una forma di glomerulopatia a depositi di C3 (Malattia a Depositi Densi o Glomerulonefrite a Depositi di C3), oppure una microangiopatia trombotica (in questo caso una forma atipica di sindrome emolitico-uremica).
- Tubulopatia prossimale da catene leggere. È caratterizzata dal tentativo di degradazione lisosomiale da parte delle cellule del tubulo prossimale di proteine monoclonali (più spesso catene leggere kappa) presenti nella pre-urina, causando un danno cellulare secondario all’accumulo intra-lisosomiale e citoplasmatico della proteina monoclonale, che quindi si deposita in forma amorfa (rara) o cristallina.
- Altre patologie da depositi organizzati: segnaliamo, seppur rare, delle forme di glomerulonefrite da immunotattoidi e fibrillari caratterizzate da depositi monoclonali di immunoglobuline. La crioglobulinemia tipo I, una forma di vasculite dei piccoli vasi secondaria a depositi organizzati di immunoglobuline, si osserva quando una proteina monoclonale assume caratteristiche chimico-fisiche per cui precipita a livello endoteliale (e spesso glomerulare) quando esposta a temperature inferiori ai 37 °C. Il quadro istologico renale è di una glomerulonefrite membrano-proliferativa a depositi e pseudo-depositi organizzati monoclonali.
- Pattern ibridi. Come prima accennato per le PGNMID, le paraproteinemie possono mimare quadri di glomerulonefrite primitiva, ad esempio di glomerulonefrite membranosa (vedi forma IgG3 kappa mediata) (6), nefropatia a prevalenti depositi di IgA (7), sindrome da anti GBM, ricorrente (8), etc. Pertanto, un attento studio di IF/IHC è necessario sul tessuto renale (preferibilmente con digestione enzimatica) al fine di distinguere un quadro di autoimmunità da una proteina monoclonale circolante, modificando radicalmente l’approccio terapeutico.
Il trattamento è diretto contro il clone plasmacellulare, B cellulare o linfoplasmacellulare responsabile della paraproteinemia. Seppur non siano presenti in molti casi delle caratteristiche di malignità del clone, il trattamento di tali condizioni è da prendere in considerazione poiché alcune condizioni mostrano una scarsa sopravvivenza renale (tabella 2).
| MGRS | N | Follow-up (months) | Kidney outcomes |
| LCDD1 | 63 | 28 | 57% ESRD |
| PGNMID2 | 37 | 30 | 38% Complete/partial recovery 38% Persistent renal dysfunction 22% ESRD |
| ITGN3 | 16 | 48 | 50% Remission 33% Persistent renal dysfunction 17% ESRD |
| MIDD4 | 64 | 25 | 39% ESRD 57% Stable/improved renal function |
| LCDD5 | 53 | 74 | 53% ESRD (10% ESRD at presentation) |
1) Pozzi C et al. Am J Kidney Dis. 2003
2) Nasr SH et al. J Am Soc Nephrol. 2009
3) Nasr SH et al. Nephrol Dial Transplant. 2012
4) Nasr SH et al. Clin J Am Soc Nephrol 2012
5) Sayed RH et al. Blood 2015
Tabella 2 – Mod. da (9)
Bibliografia
- Blade J, Fernandez-Llama P, Bosch F, Montoliu J, Lens XM, Montoto S, et al. Renal failure in multiple myeloma: presenting features and predictors of outcome in 94 patients from a single institution. Arch Intern Med. 1998;158(17):1889-93.
- Leung N, Bridoux F, Hutchison CA, Nasr SH, Cockwell P, Fermand JP, et al. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood. 2012;120(22):4292-5.
- Merlini G, and Stone MJ. Dangerous small B-cell clones. Blood. 2006;108(8):2520-30.
- Bridoux F, Leung N, Hutchison CA, Touchard G, Sethi S, Fermand JP, et al. Diagnosis of monoclonal gammopathy of renal significance. Kidney Int. 2015;87(4):698-711.
- Zand L, Kattah A, Fervenza FC, Smith RJ, Nasr SH, Zhang Y, et al. C3 glomerulonephritis associated with monoclonal gammopathy: a case series. Am J Kidney Dis. 2013;62(3):506-14.
- Debiec H, Hanoy M, Francois A, Guerrot D, Ferlicot S, Johanet C, et al. Recurrent membranous nephropathy in an allograft caused by IgG3kappa targeting the PLA2 receptor. J Am Soc Nephrol. 2012;23(12):1949-54.
- Soares SM, Lager DJ, Leung N, Haugen EN, and Fervenza FC. A proliferative glomerulonephritis secondary to a monoclonal IgA. Am J Kidney Dis. 2006;47(2):342-9.
- Fervenza FC, Terreros D, Boutaud A, Hudson BG, Williams RA, Jr., Donadio JV, Jr., et al. Recurrent Goodpasture’s disease due to a monoclonal IgA-kappa circulating antibody. Am J Kidney Dis. 1999;34(3):549-55.
- Hogan JJ, Mocanu M, Berns JS. The Native Kidney Biopsy: Update and Evidence for Best Practice. Clin J Am Soc Nephrol. 2016;11(2):354–362